Xing Lu1,2,
Xing-Lin Zhang2,
Kai Chu2,
Guo-Dong Zhang2,
Yan-Ping Zheng1 ![]()
For correspondence:- Yan-Ping Zheng Email: zhangyanpingzyp@hotmail.com Tel:+8653182169114
Received: 12 July 2015 Accepted: 29 December 2015 Published: 28 February 2016
Citation: Lu X, Zhang X, Chu K, Zhang G, Zheng Y. Targeting integrin-ß1 impedes cytokine-induced osteoclast differentiation: a potential pharmacological intervention in pathological osteolysis. Trop J Pharm Res 2016; 15(2):299-305 doi: 10.4314/tjpr.v15i2.11
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To examine whether integrin-β1 is essential for osteoclast differentiation and function and if it can be targeted for pharmacological intervention in pathological osteolysis.
Methods: Control and Integrin-β1 knockdown RAW 264.7 cells were treated with receptor activator of nuclear factor kappa-B (RANKL) or TNF-α and evaluated for osteoclast differentiation. Osteoclast differentiation and function were evaluated by marker protein analysis, tartrate-resistant acid phosphatase (TRAP) and resorption assays. Furthermore, downstream molecular signaling analysis was probed using small molecule inhibitors and blocking antibodies, and evaluated by immunoblotting.
Results: Integrin-β1 knockdown cells showed reduced osteoclast differentiation following TNF-α treatment while no change was seen after RANKL treatment (p < 0.05). Immunoblot-based molecular signaling analysis showed involvement of MAPK kinase signaling in mediating TNF-α/integrin-β1-induced osteoclastogenesis. Finally, when MAPK kinase inhibitor (2.5 and 5 µM; p < 0.05) and integrin-β1 blocking antibody (2.5 and 5 µg/mL; p < 0.05) was used to specifically attenuate TNF-α induced osteoclastogenesis, no change was observed in RANKL-induced osteoclast formation.
Conclusion: The data obtained highlight the role of integrin-β1 in TNF-α-induced osteoclastogenesis, but not in RANKL pathway. Given that, inflammatory cytokine secretions such as TNF-α are progressively implicated in pathological osteolysis, targeting this pathway may attenuate osteolysis in pathological bone tissues.
Introduction
Bone tissue changes are largely regulated by the combined action of two bone cell types - osteoblasts and osteoclasts. Osteoblasts are primarily responsible for new bone formation, while osteoclasts are responsible for removing mineralized tissue [1]. Their formation and activity are tightly regulated by a combinatorial action of multiple hormones and cytokines within the bone microenvironment [1,2]. Disturbance in these cells –altered mass and activity underlies many pathological bone conditions [3,4]. In particular, most adult skeletal disease such as osteoarthritis, rheumatoid arthritis is related to abnormal osteoclast activity [3,4]. Pro-inflammatory secretions such as TNF-α, IL-6 produced in the pathological bone tissues have been implicated to progressively modulate the osteoclast activity, facilitating the disease progression, and hence targeting their involvement has been suggested as an intervention to reduce bone erosion [5,6].
Studies from targeted knockout mutations or other naturally occurring mutations have identified at least 24 gene loci to regulate osteoclast differentiation and activation [7,8]. However, recent advance have underscored the importance of the tumor necrosis factor (TNF) receptor (TNFR)/TNF-like proteins: (a) osteoprotegerin (OPG), (b) receptor activator of nuclear factor (NF)-ĸB (RANK) and (c) RANK ligand (RANKL) to regulate osteoclast function. It is now known that RANKL or TNF-α combined with macrophage colony stimulating factor (MCSF) alone is sufficient to induce osteoclastogenesis from bone marrow macrophage cells in vitro [9]. However, deeper understanding of how diverse physiological and pathological signals modulate the RANKL or TNF-α pathway and osteoclast functions would allow intervening therapeutics to regulate bone erosion.
Integrins are heterodimeric receptors mediating cell-cell and cell-matrix interactions and a role for them has been widely appreciated in the osteoclast differentiation and activation [10,11]. Expression analyses of osteoclast have identified at least three major integrin types – alpha V beta 3 (α5β3), alpha 5 beta1 (α5β1), alpha 2 beta1 (α2β1) [12]. Although a critical role for α5β3 integrin has been defined in osteoclastogenesis [10], the other integrin subtypes remain largely unexplored. Given that most of them involve integrin-β1, understanding the functions through integrin-β1 would allow understanding the functions of both the integrin subtypes and other functions involving integrin-β1 in osteoclasts.
In this study, the role of integrin subtype – integrin-β1 has been highlighted to specifically regulate the TNF-α induced osteoclast formation and activation by modulating MAPK signaling. In addition, blocking this pathway through integrin-β1 blocking antibody or MAPK inhibitor showed similar results and thereby provide a viable route for therapeutic intervention.
Methods
RNAi
Mouse monocyte cells RAW 264.7 were purchased from American Type Culture Collection. Cells were maintained in DMEM supplemented with 10 % FBS, 1 mM pyruvate and 1 % pen-strep solution (Life Technologies, USA). shRNA sequences targeting integrin-β1 were cloned into pLKO.1 vector and virus were produced in 293T cells by transient transfection of the lentiviral vector (pLKO.1) and packaging vectors (pPAX2 and pMD2) for 48 h. Control and RNAi virus were then concentrated by ultracentrifugation and infected in RAW 264.7. Virus infected cells were then selected for puromycin and evaluated for integrin-β1 knockdown efficiency.
Osteoclast differentiation
For differentiation, control vector and integrin-β1 knockdown RAW264.7 cells were plated with a density of 5000 cells/cm2 and supplemented with either RANKL (50 ng/mL) or TNF-alpha (50 ng/mL) combined with M-CSF (100 ng/mL) (Sigma-Aldrich, USA) for 7 days with media changes after every two days. Seven days of culture has been used based on the previous observation (unpublished data) of the minimum period required to yield significant osteoclast differentiation with the used conditions.
TRAP staining
RANKL or TNF-alpha treated culture systems were fixed in 10 % buffered formalin and stained for TRAP (Sigma-Aldrich, USA). Presence of three or more nuclei was used as criteria to identify osteoclasts.
Osteoclastic resorption
Osteoclast differentiation was induced in osteoassay plates (Corning, USA) for 1 week and the cells were removed with 10 % NaClO, washed with water and air-dried. Resorption pits were measured using PixeLINK Capture SE software.
RT-PCR
Total RNA was isolated from 1-week differentiation cultures and reverse transcribed as described previously [13]. PCR was performed with a 7500 Applied Biosystem instrument using TaqMan probes with the universal PCR Master Mix (Life Technologies, USA). The following probes were used: Taqman: Ctsk (Cathepsin K): Mm00484039_m1; Sp7 (Osterix): Mm00504574_m1; GAPDH (glyceraldehyde 3-phosphate dehydrogenase): Mm99999915_g1 (Applied Biosystems, USA). Untreated samples were used as reference to determine the changes in gene expression.
Immunoblotting
Immunoblotting for proteins were performed using standard procedures as described previously [13]. Briefly, the cells were lysed using a RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 % Nonidet P-40, 1 mM EDTA, 10 mg/ml aprotinin, 1 mg/ml leupeptin, 0.1 M phenylmethylsulfonyl fluoride, 0.5 M sodium fluoride and 1 mM sodium orthovanadate). Equal amount of proteins were separated by 4-20 % gel under reducing conditions, transferred to a nitrocellulose membrane (Bio-Rad, #162-0115; 0.45 µm) and blotted using specific antibodies for proteins described in the proteins (Cell Signaling Technology, USA). The membrane was then incubated with respective secondary antibodies and developed using Super Signal West Dura (Pierce Biotechnology, USA).
Inhibition assay
Cells were incubated with varying concentrations of blocking integrin-β1 antibody (Millipore, USA) or MAPK kinase inhibitor (PD098059, Alexis, UK) along with respective RANKL or TNF-alpha treatments. After differentiation, cells were probed for osteoclasts using TRAP assay.
Statistics
All statistical computing and graphical presentations were carried out using GraphPad Prism Software (Version 6.0) (GraphPad Inc, CA, USA). Data are presented as mean ± standard deviation (SD) with sample size (n) indicating the number of independent experiments. Differences were assessed by ANOVA and accepted as statistically significant at p < 0.05.
Results
Characteristics of stable Integrin beta -1 knockdown cells
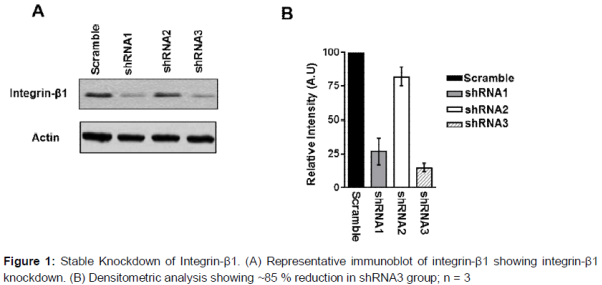
The shRNA sequences targeted for integrin-β1 were adapted from vendor sources (validated clones from Sigma RNAi consortium) and also previously used and validated elsewhere [13]. shRNA sequences were cloned into lentiviral vector (pLKO.1) and infected in RAW264.7. The cells were then selected with puromycin and evaluated for knockdown efficiency. Cells transduced with empty vector showed similar integrin-β1 expression levels as normal cells, whereas cells transduced with shRNA sequences had varying levels of reduction (A and B). Cells with the greatest reduction of integrin-β1 (~85 % reduction) were designated as integrin-β1 knock down cells and used in subsequent experiments to mimic the loss of gene function phenotype.
Integrin-β1 is essential for osteoclast differentiation
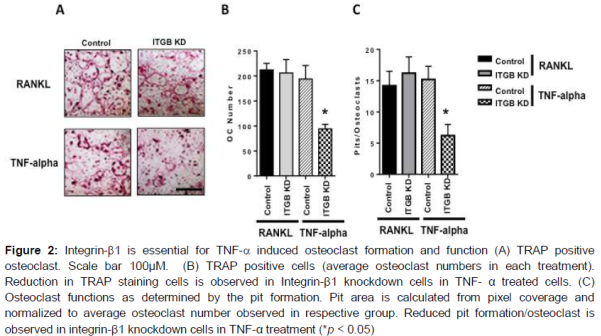
To evaluate whether integrin-β1 is essential for osteoclast differentiation, we examined the effects of loss of integrin-β1 function in vitro osteoclast differentiation. Stable knockdown of integrin-β1 strongly decreased the formation of TRAP positive osteoclasts in TNF-α induced osteoclastogenesis, while RANKL treated cells differentiated normally (A and B). These observations were further evaluated for functional significance through pit formation assay.
In accordance with the osteoclast formation trends, integrin-β1 knockdown cells showed reduced pit formation upon TNF-α treatments and no difference were seen in the RANKL treatments (C).
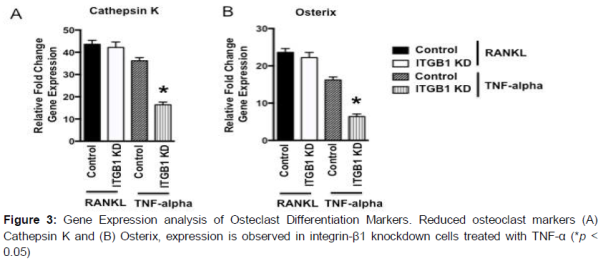
Finally, the results were confirmed by the decreased expression of osteoclast differentiation marker genes such as Osterix and Cathespin k in integrin-β1 knockdown cells in TNF-α treatments, and no change in RANKL treatments were observed (A and B). Together, these results suggest that the integrin-β1 related signaling plays a critical role in TNF-α induced osteoclast differentiation.
MAPK kinase signaling is associated with osteoclast differentiation
To elucidate the molecular mechanisms relating integrin-β1 with osteoclastogenesis, we evaluated for changes in signaling cascades associated with osteoclast differentiation. We compared the changes between control and integrin-β1 knockdown cells upon treatment with either RANKL or TNF-α.
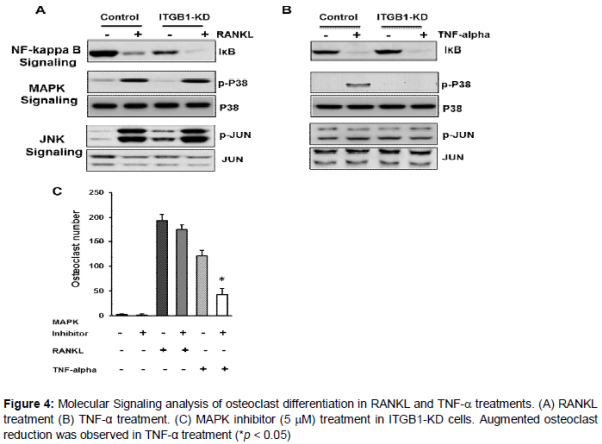
In response to TNF-α stimulation, loss of integrin-β1 showed reduced p38 MAP kinase phosphorylation, while no differences were seen in the phosphorylation status of JNK signaling, and IĸB levels between control and integrin-β1 knockdown cells (A and B).
In contrast, upon RANKL stimulation no significant changes were seen between control and integrin-β1 knockdown cells in any of the signaling pathways investigated including p38 (A and B). However, all the pathways investigated showed increased response to RANKL. To further confirm the specificity of Integrin-β1 and p38 MAP kinase signaling in relation to osteoclast differentiation, the integrin-β1 knockdown cells were treated with p38 MAP kinase inhibitor and an augmented reduction in osteoclast differentiation were observed in integrin-β1 knockdown cells only in TNF-α treatments (C). Surprisingly, though increased p38 response was seen in RANKL treatment, p38 inhibitor does not show a significant reduction in osteoclast differentiation. Although speculative, this implies that the significance of MAPK kinase signaling is minimal in RANKL induced osteoclasts, which is in contrast to TNF-α induced osteoclast formation (C). Together, this data confirms that integrin-β1 is critical and functions via MAP kinase signaling in TNF-α induced osteoclast differentiation.
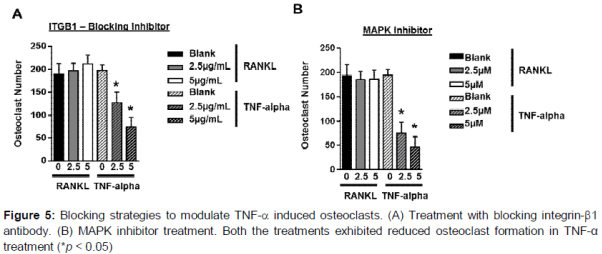
Integrin-β1 blocking antibody and MAPK kinase inhibitor prevents TNF-α mediated osteoclast differentiation
Based on the observation for a role for integrin-β1 in mediating osteoclast differentiation, we further investigated whether the integrin-β1 blocking antibodies could modulate the signaling and osteoclast formation. When control cells were continuously treated with increasing doses of integrin-β1 blocking antibody, the formation of TRAP multinucleated cells upon TNF-α treatment was attenuated in a dose dependent manner, while no significant changes were seen in RANKL treated cells (A). Similar results were observed upon MAPK kinase inhibitor treatments in TNF-α treatments and no change was seen in RANKL treated cells (B).
Discussion
The significance of ODF/RANKL-RANK pathway in osteoclast formation has been extensively studied in various model systems [7,8,14]. However, the significance of other pathways such as TNF-a induced osteoclast formation remains largely unexplored. In addition molecular cross talk between TNF-α and RANKL signaling also add to the complexity and make it difficult to segregate individual pathway components [15]. Given that, TNF-a and other pro-inflammatory cytokines are present in excess in pathological skeletal tissues exclusively, understanding and targeting the pathway could specifically attenuate bone erosion in pathological skeletal tissues and thereby make it an attractive target for pharmacological interventions. In this study, we have shown that integrin-b1 is essential for the TNF-a induced TRAP positive cells formation, but not for the RANKL induction based osteoclasts. Gene expression analysis of osteoclast markers in integrin-b1 knockdown cells has also confirmed similar observation of reduced expression in TNF-a treated cells and not in RANKL treated cells, implying for a specific role of integrin-b1 in TNF-a induced osteoclasts.
Determination of signaling events following TNF-a induction and the involvement of integrin-b1 in it is a critical to decipher and until now no consensus for exclusive downstream signaling event of TNF-a induction in osteoclastogenesis is attained [15]. However, mounting evidence have highlighted a signaling cross talk between TNF-a pathway and integrin signaling pathways [2,3,16]. In addition, earlier reports to understand the osteoclastogenesis have also suggested for a role for JNK, NF-kB, p38 MAPK signaling pathway in osteoclast formation and activation [15,17]. In accordance changes in IkB and p38 levels were observed upon TNF-a induction, while it is lost for only p38 in integrin-b1 knockdown cells. This suggests that TNF-a induced TRAP positive cells are mediated by integrin-b1 via p38 signaling. In addition, we also tested whether other pro-inflammatory cytokines could also have such effects and our result with IL-1 induction though showed IkB reduction, didn’t show any change in p38 or any effect on TRAP positive cells (data not shown). In accordance, IL-1 is also known for other molecule dependence to modulate osteoclast activity [18]. On the other hand, RANKL treatments showed complete independence of integrin-β1 over all the pathways tested. This suggests that TNF-α is a critical cytokine among pro-inflammatory molecules in pathological tissues to modulate osteoclast formation and activity and integrin-b1/p38 is specifically involved in the TNF-a induced osteoclast formation.
Previous studies targeting integrins to attenuate the osteoclast functions have largely focused on αvβ3 integrins [19]. However, lack of specificity to disease bone tissues limits its translation. Given that integrin-β1 is associated with TNF-α induction, targeting integrin-β1/p38 pathway could specifically attenuate the osteolytic condition in pathological tissues. Earlier report to attenuate the integrin-β1 has used antisense oligonucleotides technology [20], but lack of efficient delivery methods for DNA oligonucleotides warrants for other small molecule based methods to attenuate osteoclast activity. Our antibody or small molecule inhibitor based approaches could provide alternate systems and in accordance our in vitro data have also provided the evidence to attenuate osteoclast activity. However, future studies for in vivo validation and evaluation of off target effects is warranted.
Conclusion
Overall, the data presented in this study underscores the critical role of integrin-β1 in TNF-α-induced osteoclast formation and resorption, and its potential to reduce pathological osteolysis. Future studies validating the role of integrin-β1 in osteoclast maturation and activation from in vivo pathological bone tissues, and improvements in developing efficient antibody-based therapeutic options or other alternate routes to target the pathway could translate into a viable management option for chronic bone disease.
References
Archives


News Updates